3-C. Machine Learning Models
3.3 Graph Neural Networks for Molecular Data
Graph neural networks (GNNs) are useful in chemistry because molecules are already graph-like objects. A molecule is made of atoms connected by bonds. A molecular GNN keeps that structure instead of flattening the molecule into one fixed list of descriptors at the start.
The goal of this section is to build the idea one piece at a time:
- What a molecular graph is.
- What data a GNN receives.
- How this differs from molecular fingerprints.
- How message passing works.
- How atom-level information becomes one molecule-level prediction.
- How simple GCN and MPNN models are implemented.
The complete runnable notebooks can still be used for practice:
- Completed and Compiled Code (3.3.1-3.3.3): Click Here
- Completed and Compiled Code (3.3.4): Click Here
3.3.1 What Is a Molecular Graph?
A graph is a set of nodes connected by edges. In a molecular graph:
- one molecule is represented as one graph
- atoms are the nodes
- bonds are the edges
For ethanol, CH3-CH2-OH, the heavy-atom graph is:
C0 - C1 - O2
Here C0 is the carbon in CH3, C1 is the carbon in CH2, and O2 is the oxygen in OH. The hydrogens are often treated as implicit because many molecular datasets store only heavy atoms by default.
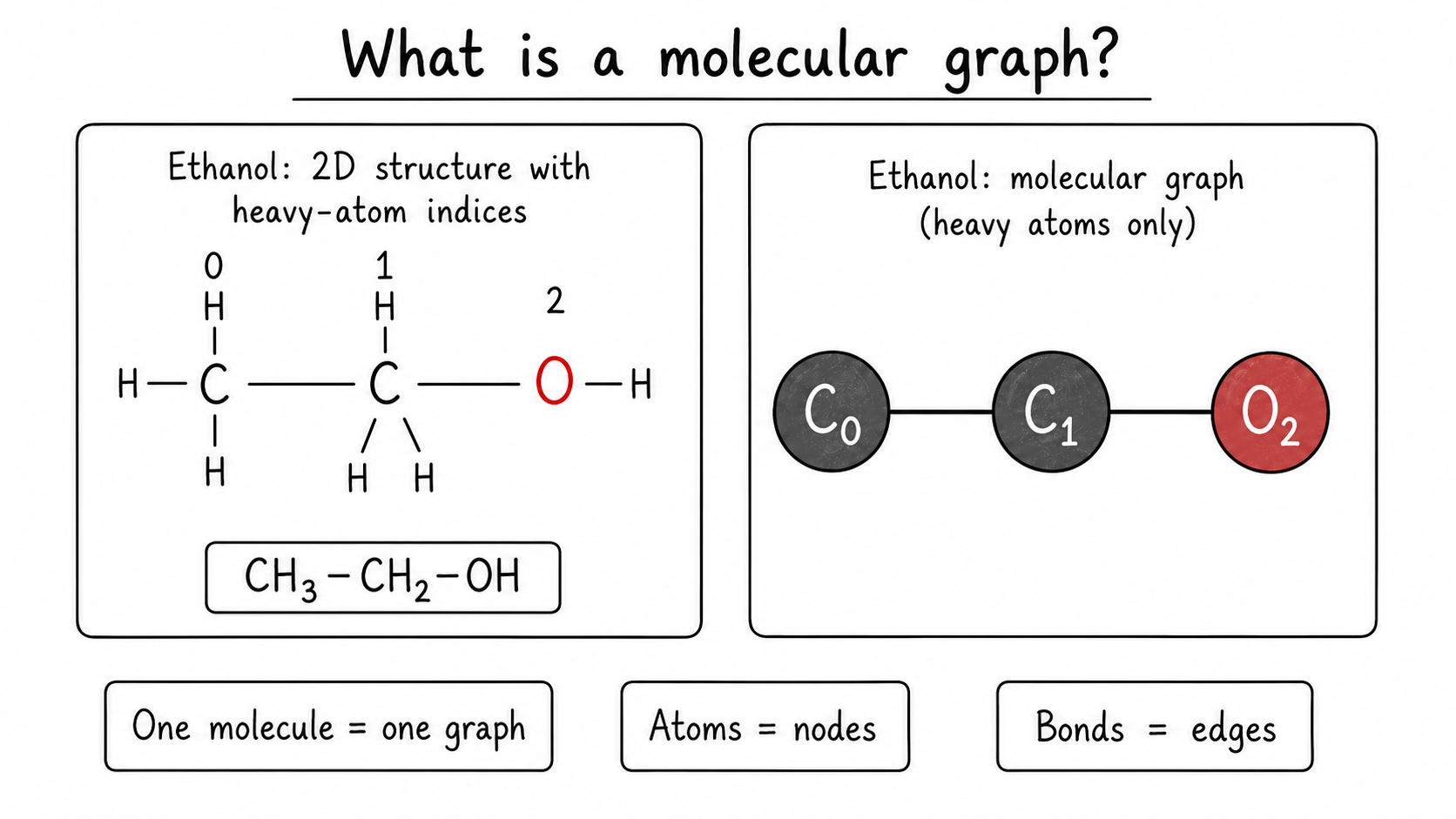
Code for this idea. RDKit can read a SMILES string and expose the atoms and bonds that become the graph.
from rdkit import Chem
smiles = "CCO" # ethanol: CH3-CH2-OH
mol = Chem.MolFromSmiles(smiles)
print("Atoms")
for atom in mol.GetAtoms():
print(atom.GetIdx(), atom.GetSymbol())
print("\nBonds")
for bond in mol.GetBonds():
start = bond.GetBeginAtomIdx()
end = bond.GetEndAtomIdx()
print(start, "-", end, bond.GetBondType())
Expected output:
Atoms
0 C
1 C
2 O
Bonds
0 - 1 SINGLE
1 - 2 SINGLE
This is the first important shift: the model is not looking at a drawing of ethanol. It receives atoms, bonds, and numerical features derived from them.
3.3.2 What Information Is Stored in a Molecular Graph?
A molecular graph needs more than connectivity. The model also needs information about each atom and each bond.
Common atom features include:
- element type, such as C, N, O, or Cl
- formal charge
- aromaticity
- hybridization
- number of attached hydrogens
Common bond features include:
- single, double, triple, or aromatic bond type
- whether the bond is conjugated
- whether the bond is in a ring
In PyTorch Geometric, this information is usually stored in three objects:
x: the node feature matrixedge_index: which atoms are connectededge_attr: bond features for each edge
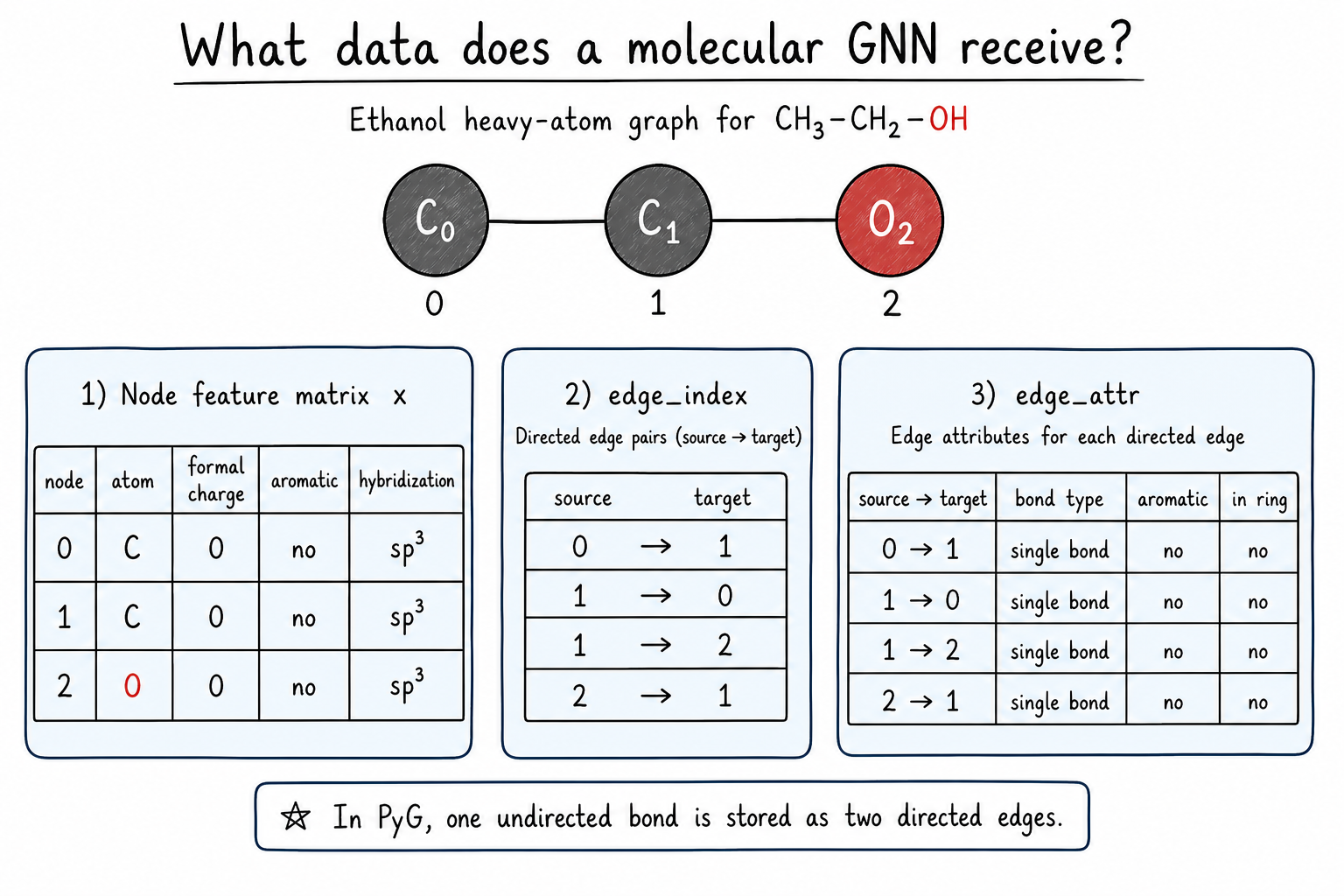
Code for this idea. This example builds a small PyTorch Geometric Data object from ethanol. The features are deliberately simple so that the representation is easy to inspect.
import torch
from rdkit import Chem
from torch_geometric.data import Data
mol = Chem.MolFromSmiles("CCO")
atom_features = []
for atom in mol.GetAtoms():
atom_features.append([
atom.GetAtomicNum(), # C=6, O=8
atom.GetFormalCharge(),
int(atom.GetIsAromatic()),
int(atom.GetHybridization() == Chem.HybridizationType.SP3),
])
edge_index = []
edge_features = []
for bond in mol.GetBonds():
i = bond.GetBeginAtomIdx()
j = bond.GetEndAtomIdx()
is_single = int(bond.GetBondType() == Chem.BondType.SINGLE)
is_aromatic = int(bond.GetIsAromatic())
is_in_ring = int(bond.IsInRing())
# Store each undirected bond as two directed edges.
edge_index += [[i, j], [j, i]]
edge_features += [
[is_single, is_aromatic, is_in_ring],
[is_single, is_aromatic, is_in_ring],
]
x = torch.tensor(atom_features, dtype=torch.float)
edge_index = torch.tensor(edge_index, dtype=torch.long).t().contiguous()
edge_attr = torch.tensor(edge_features, dtype=torch.float)
data = Data(x=x, edge_index=edge_index, edge_attr=edge_attr)
print(data)
One detail matters for PyG: an ordinary chemical bond is undirected, but edge_index stores message directions. A C-C bond is therefore written as both 0 -> 1 and 1 -> 0.
The table in the figure is human-readable. In a real model, symbols such as C, O, and sp3 must be encoded as numbers before training.
3.3.3 How Is This Different From a Fingerprint?
A molecular fingerprint is a fixed-length vector. It records whether certain substructures are present, but it does not keep the molecule as an atom-bond network during learning.
A molecular graph keeps the molecule connected. The model can update each atom by looking at its bonded neighbors. This is the main practical difference:
- A regular neural network usually receives one fixed vector per molecule.
- A GNN receives a graph: atom features plus bond connections.
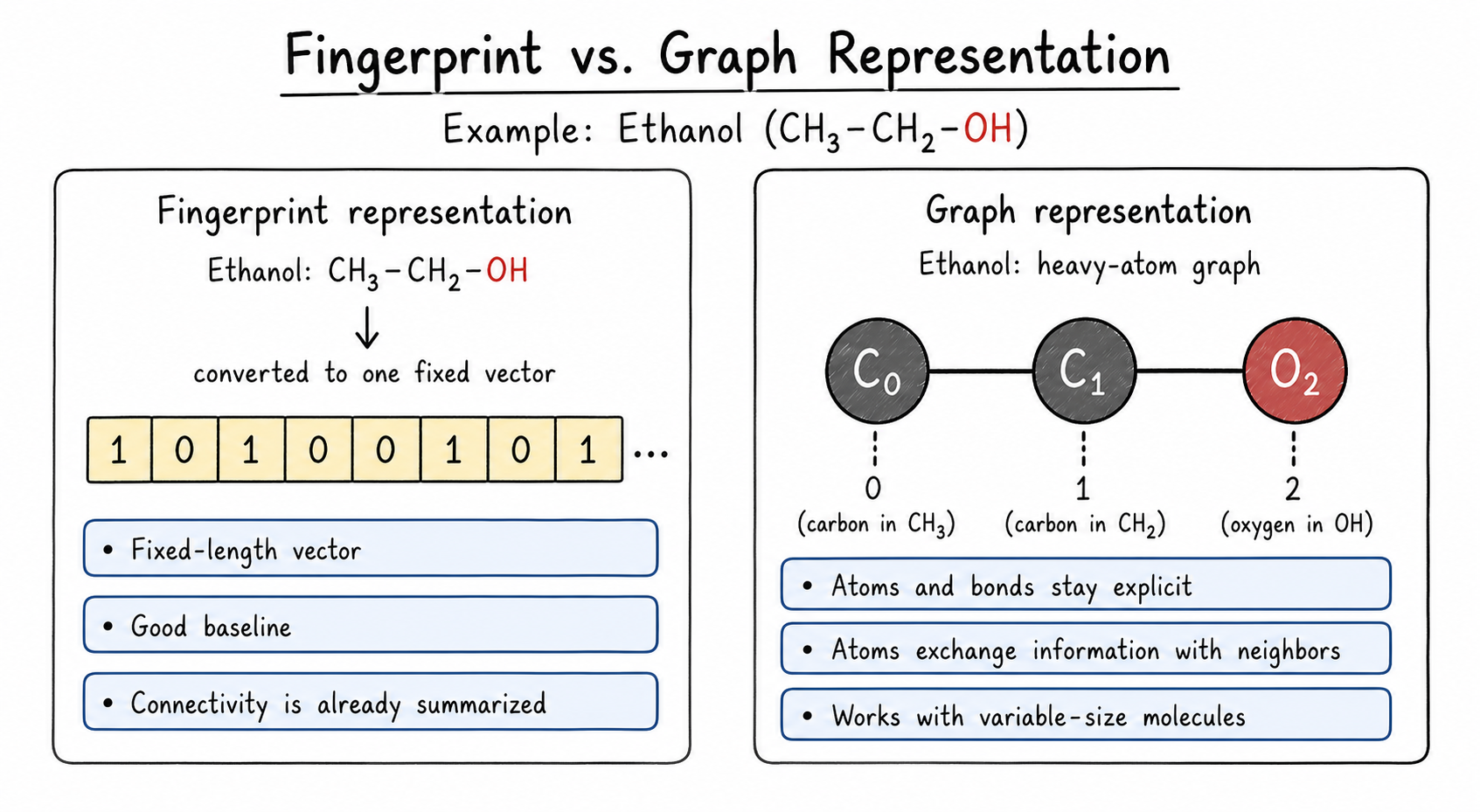
Code for this idea. The same SMILES string can be converted into either a fingerprint or a molecular graph.
from rdkit import Chem
from rdkit.Chem import rdFingerprintGenerator
mol = Chem.MolFromSmiles("CCO")
# Fingerprint: one fixed vector for the whole molecule.
morgan = rdFingerprintGenerator.GetMorganGenerator(radius=2, fpSize=128)
fp = morgan.GetFingerprint(mol)
fp_bits = list(fp.ToBitString())
print("Fingerprint length:", len(fp_bits))
# Graph: atoms and bonds remain explicit.
print("Number of atoms:", mol.GetNumAtoms())
print("Number of bonds:", mol.GetNumBonds())
Fingerprints are still very useful. They are fast, simple, and often strong baselines. GNNs become attractive when we want the model to learn directly from atom-bond connectivity instead of relying only on precomputed molecular descriptors.
3.3.4 What Is a Graph Neural Network?
A graph neural network updates each node by using information from nearby nodes. In a molecular GNN, that means each atom updates its representation using information from bonded atoms.
What actually gets trained? The molecular graph is the input, not the part that learns. The trainable part is the neural-network math that acts on that graph: the message-passing layers learn how to transform information from neighboring atoms, and the final prediction layer learns how to turn the molecule representation into a property prediction.
| Part of the pipeline | Role |
|---|---|
| Atom features, such as atom type, charge, aromaticity | Input data; not trained |
| Graph connectivity, such as which atoms are bonded | Input structure; not trained |
| Graph convolution / message passing | Trainable math; updates atom representations |
| Atom embeddings | Intermediate representations produced by the model |
| Pooling / readout | Usually a fixed combine step, such as mean or sum |
| Final prediction layer | Trainable math; maps the molecule vector to the target property |
At the start, an oxygen atom may only know simple facts like “I am oxygen” and “I am sp3.” After one message-passing layer, it can know that it is attached to carbon. After two layers, it can receive information from atoms two bonds away. In ethanol, the oxygen can gradually learn that it is part of an alcohol attached to an ethyl group.
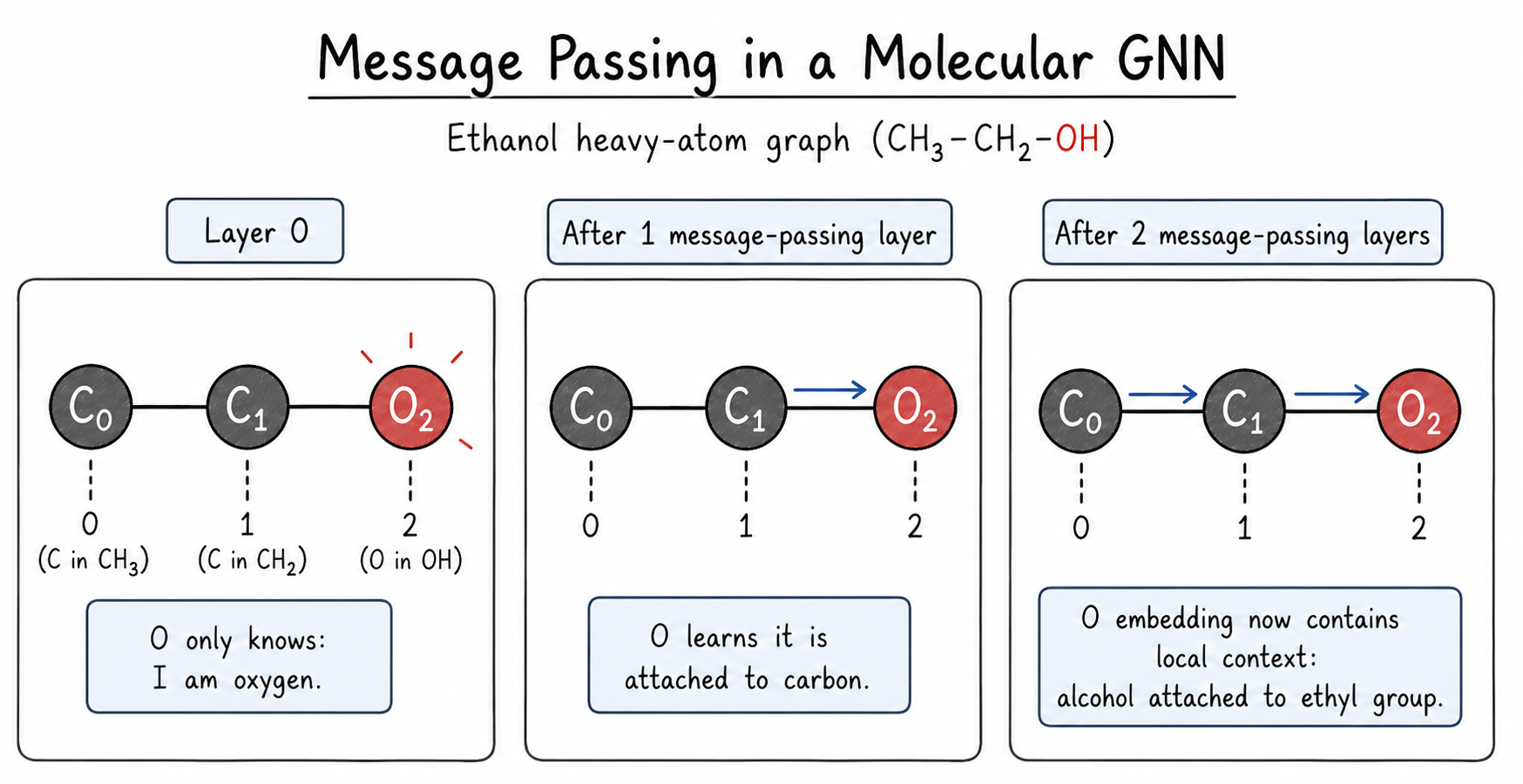
The main operation is:
- Send messages. Neighboring atoms send information along bonds.
- Aggregate messages. Each atom collects messages from its neighbors.
- Update the atom. The model combines the old atom information with the new neighbor information.
Code for this idea. The following toy code shows message passing without hiding the idea inside a library layer.
import torch
import torch.nn as nn
# Three atoms: C0, C1, O2.
# Here each atom has a tiny feature vector.
h = torch.tensor([
[6.0, 0.0], # C0
[6.0, 0.0], # C1
[8.0, 0.0], # O2
])
# Directed edges: 0->1, 1->0, 1->2, 2->1.
edge_index = torch.tensor([
[0, 1, 1, 2],
[1, 0, 2, 1],
])
message_layer = nn.Linear(2, 2)
update_layer = nn.Linear(4, 2)
messages = torch.zeros_like(h)
for src, dst in edge_index.t():
messages[dst] += message_layer(h[src])
h_next = torch.relu(update_layer(torch.cat([h, messages], dim=1)))
print(h_next)
This is not a full chemical model. It is only meant to show the mechanism: each atom receives transformed information from its bonded neighbors.
3.3.5 How Does a GNN Make One Molecular Prediction?
After message passing, every atom has an updated representation. But many chemistry tasks need one prediction for the whole molecule, such as:
- solubility
- toxicity
- binding activity
- blood-brain barrier permeability
So the model must combine all atom representations into one molecule representation. This step is called pooling or readout.
For chemists, the simplest way to read “pooling” is:
Pooling means summarizing all atom-level information into one molecule-level representation.
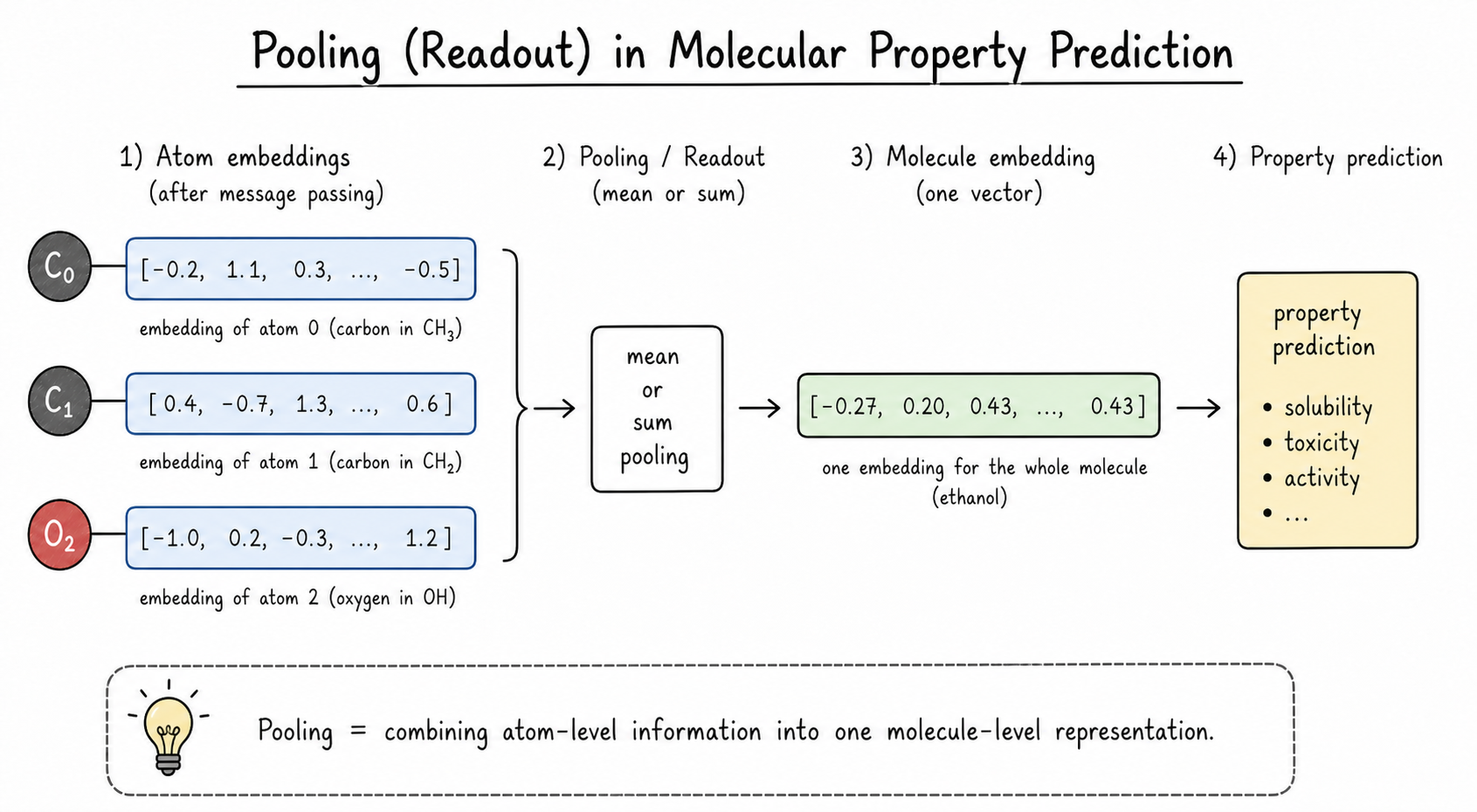
Code for this idea. PyG provides pooling functions that know which atoms belong to which molecule in a batch.
import torch
from torch_geometric.nn import global_mean_pool, global_add_pool
# Suppose message passing produced one embedding per atom.
atom_embeddings = torch.tensor([
[0.2, 1.1, 0.4], # atom 0
[0.5, 0.7, 0.3], # atom 1
[1.3, 0.2, 0.9], # atom 2
])
# All three atoms belong to molecule 0.
batch = torch.tensor([0, 0, 0])
molecule_mean = global_mean_pool(atom_embeddings, batch)
molecule_sum = global_add_pool(atom_embeddings, batch)
print("Mean pooled molecule vector:", molecule_mean)
print("Sum pooled molecule vector:", molecule_sum)
The molecule vector is then passed to a small neural network layer that produces the final prediction.
3.3.6 A Simple GCN Model
A Graph Convolutional Network (GCN) is one of the simplest practical GNNs. A GCN layer updates each atom by mixing information from neighboring atoms. It treats the graph structure as the guide for where information can flow.
GCNs are useful as a first model because they are short and easy to inspect. They do not use detailed bond features as strongly as an MPNN, but they show the basic GNN workflow clearly.
Code for this idea. This model takes a PyG Data object, updates atom embeddings with GCN layers, pools the atom embeddings into a molecule embedding, and predicts one value.
import torch.nn as nn
import torch.nn.functional as F
from torch_geometric.nn import GCNConv, global_mean_pool
class MolecularGCN(nn.Module):
def __init__(self, node_features, hidden_dim=64):
super().__init__()
self.conv1 = GCNConv(node_features, hidden_dim)
self.conv2 = GCNConv(hidden_dim, hidden_dim)
self.predict = nn.Linear(hidden_dim, 1)
def forward(self, data):
x = data.x.float()
edge_index = data.edge_index
batch = data.batch
x = F.relu(self.conv1(x, edge_index))
x = F.relu(self.conv2(x, edge_index))
molecule_vector = global_mean_pool(x, batch)
return self.predict(molecule_vector).view(-1)
The important pattern is:
atom features -> graph convolution -> atom embeddings -> pooling -> prediction
For molecular property prediction, this is the first complete GNN pipeline.
3.3.7 MPNN: Using Bond Information During Message Passing
A Message Passing Neural Network (MPNN) is a broader and more chemistry-aware version of the same idea. The key difference is that an MPNN can use bond features while messages are being sent.
This matters chemically. A C-C single bond, a C=O double bond, and an aromatic bond should not always transmit information in the same way. Bond type, conjugation, and ring membership can affect the message passed from one atom to another.
In the MPNN below, NNConv uses edge_attr to shape the message along each bond.
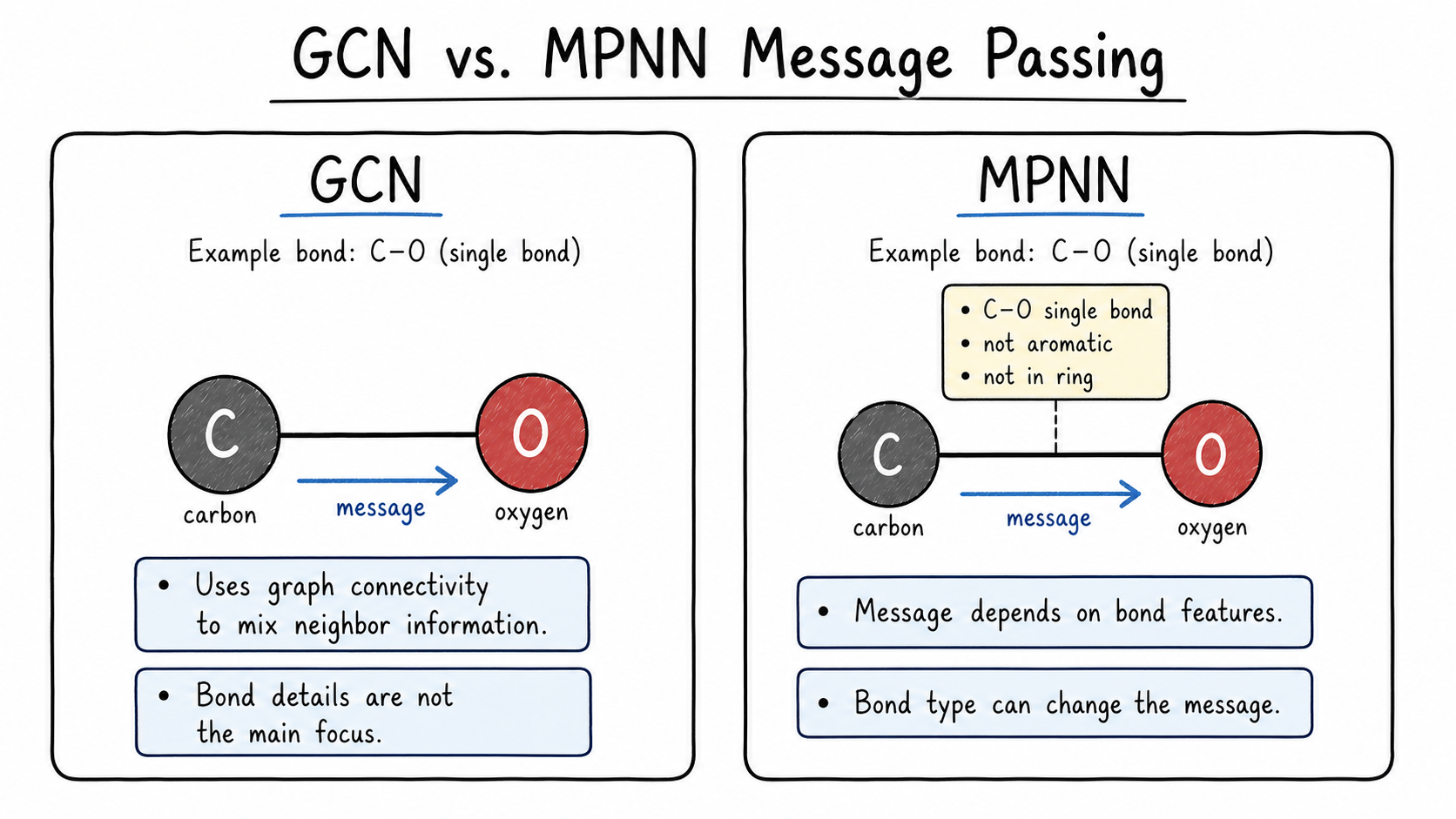
Code for this idea.
import torch
import torch.nn as nn
import torch.nn.functional as F
from torch_geometric.nn import NNConv, global_add_pool
class EdgeNetwork(nn.Module):
def __init__(self, edge_features, hidden_dim):
super().__init__()
self.net = nn.Sequential(
nn.Linear(edge_features, hidden_dim * hidden_dim),
nn.ReLU(),
nn.Linear(hidden_dim * hidden_dim, hidden_dim * hidden_dim),
)
def forward(self, edge_attr):
return self.net(edge_attr)
class MolecularMPNN(nn.Module):
def __init__(self, node_features, edge_features, hidden_dim=64, layers=3):
super().__init__()
self.atom_embed = nn.Linear(node_features, hidden_dim)
self.edge_net = EdgeNetwork(edge_features, hidden_dim)
self.convs = nn.ModuleList([
NNConv(hidden_dim, hidden_dim, self.edge_net, aggr="add")
for _ in range(layers)
])
self.gru = nn.GRU(hidden_dim, hidden_dim)
self.predict = nn.Linear(hidden_dim, 1)
def forward(self, data):
x = self.atom_embed(data.x.float())
edge_index = data.edge_index
edge_attr = data.edge_attr.float()
batch = data.batch
h = x.unsqueeze(0)
for conv in self.convs:
message = F.relu(conv(x, edge_index, edge_attr)).unsqueeze(0)
out, h = self.gru(message, h)
x = out.squeeze(0)
molecule_vector = global_add_pool(x, batch)
return self.predict(molecule_vector).view(-1)
This model is more complex than the GCN, but the reason is chemical: the message is allowed to depend on the bond.
3.3.8 Training and Evaluation
Training a molecular GNN follows the same broad pattern as other neural networks:
- Convert molecules into graph data.
- Feed each graph into the model.
- Compare the prediction with the known label.
- Update the model parameters with backpropagation.
- Evaluate on validation and test molecules.
For datasets such as OGB-MOLHIV, the task is binary classification: active or inactive against HIV. The dataset is imbalanced, meaning the positive class is much smaller than the negative class. In that case, accuracy can be misleading because a model can look accurate by mostly predicting the majority class.
ROC-AUC is often used because it measures how well the model ranks positive molecules above negative molecules across many thresholds.
Code for this idea. This is the shape of a minimal training step.
import torch
import torch.nn as nn
criterion = nn.BCEWithLogitsLoss()
optimizer = torch.optim.Adam(model.parameters(), lr=1e-3)
model.train()
for data in train_loader:
data = data.to(device)
y = data.y.view(-1).float()
optimizer.zero_grad()
logits = model(data)
loss = criterion(logits, y)
loss.backward()
optimizer.step()
For imbalanced classification, the loss can include a positive-class weight:
criterion = nn.BCEWithLogitsLoss(pos_weight=pos_weight)
That weight tells the model to pay more attention to the smaller positive class.
3.3.9 Practical Notes for Chemists
GNNs are powerful, but they are not magic. The most important limitations are practical:
A 2D graph is not a full 3D molecule. Most simple molecular GNNs use atoms and bonds from a 2D graph. They may miss conformation, stereochemical details, solvent effects, or protein-ligand geometry unless those features are explicitly included.
More layers are not always better. Each message-passing layer lets atoms see farther away. But if the network is too deep, atom representations can become too similar. This is called over-smoothing. In molecular tasks, 2-4 message-passing layers are often a reasonable starting point.
Interpretation needs care. A GNN may predict that a molecule is active or toxic, but the model does not automatically explain the chemistry. Methods such as attention visualization, atom attribution, or substructure masking can help, but the explanations should be checked against chemical intuition.
Generalization depends on chemical space. If the training data contains mostly one family of compounds, the model may perform poorly on a very different family. This is not unique to GNNs, but it is especially important in molecular discovery.
3.3.10 Summary
The key idea is simple:
Molecule -> molecular graph -> message passing -> pooling -> prediction
A molecular graph keeps atoms and bonds explicit. A GNN uses that connectivity to let atoms exchange information with their bonded neighbors. After several rounds of message passing, the model pools atom-level information into one molecule-level representation and predicts a property.
GCNs are a clean starting point for learning the workflow. MPNNs extend the idea by using bond features during message passing, which often better matches chemical intuition.
Section 3.3 Quiz Questions
1) What is a node in a molecular graph?
A. A whole dataset B. A molecule C. An atom D. A prediction label
Click to show answer
Correct answer: C. In the molecular graphs used here, atoms are nodes and bonds are edges.2) For ethanol, CH3-CH2-OH, what is the heavy-atom graph?
A. C-C-O
B. C-O-C
C. O-O-C
D. one node only
Click to show answer
Correct answer: A. Ethanol has two carbon atoms and one oxygen atom connected as `C-C-O`.3) Why does PyG often store one chemical bond as two edges?
A. Because the molecule has two bonds B. Because messages can be passed in both directions C. Because every atom must have two labels D. Because GNNs cannot use undirected graphs
Click to show answer
Correct answer: B. A C-C bond can send information from atom 0 to atom 1 and from atom 1 to atom 0.4) What does pooling mean in molecular property prediction?
A. Removing all atom features B. Combining atom embeddings into one molecule embedding C. Converting a molecule into a SMILES string D. Sorting molecules by molecular weight
Click to show answer
Correct answer: B. Pooling summarizes atom-level information into one molecule-level representation.5) Why might an MPNN be more chemically expressive than a simple GCN?
A. It never needs training B. It can use bond features during message passing C. It ignores atom connectivity D. It only works for ethanol
Click to show answer
Correct answer: B. Bond type, aromaticity, conjugation, and ring membership can affect the messages passed between atoms.6) Why is ROC-AUC often used for OGB-MOLHIV?
A. The dataset is imbalanced B. The molecules have no bonds C. ROC-AUC is only for regression D. Accuracy is always impossible to compute
Click to show answer
Correct answer: A. ROC-AUC is useful when the positive and negative classes are highly imbalanced.References
- Gilmer, J. et al. (2017). Neural Message Passing for Quantum Chemistry.
- Kipf, T. N., & Welling, M. (2017). Semi-Supervised Classification with Graph Convolutional Networks.
- Hu, W. et al. (2020). Open Graph Benchmark: Datasets for Machine Learning on Graphs.
- Fey, M., & Lenssen, J. E. (2019). Fast Graph Representation Learning with PyTorch Geometric.
- Sanchez-Lengeling, B. et al. (2021). A Gentle Introduction to Graph Neural Networks. Distill.